Einblick in die Orbitalstruktur organischer Halbleiter
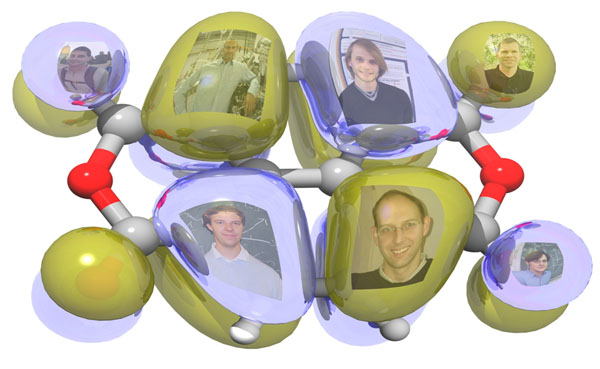
Bei großen Molekülen ist es eine wissenschaftliche Herausforderung, die Orbitale – also die Aufenthaltswahrscheinlichkeit - der Elektronen mathematisch zu berechnen. In einer Zusammenarbeit zwischen Physikern der Universitäten Würzburg und Bayreuth konnte jetzt eine geeignete Methode gefunden werden.
Molekulare Halbleiter sind das Herzstück zahlreicher neuer Technologien, beispielsweise im Bereich der Photovoltaik oder Elektronik. In der Photovoltaik eingesetzt, wandeln sie in organischen Solarzellen Licht in elektrischen Strom um. In der Elektronik leisten sie ihren Beitrag zu einer effizienteren Signalverarbeitung.
Wer die Halbleiter effizienter machen möchte, muss ihre mikroskopische Struktur möglichst genau kennen. Von besonderem Interesse sind dabei die Bereiche hoher Aufenthaltswahrscheinlichkeit – sogenannte Orbitale – der äußeren Elektronen, weil diese die physikalischen und chemischen Eigenschaften der Moleküle bestimmen. An einer mathematischen Theorie, die die Anordnung dieser Orbitale möglichst exakt beschreiben kann, hat es bislang allerdings gemangelt.
Physiker der Universität Bayreuth haben nun im Rahmen der sogenannten Dichtefunktionaltheorie ein Konzept entwickelt, das es erlaubt, die energetische Reihenfolge der Molekülorbitale mit hoher Genauigkeit zu berechnen. Die experimentelle Bestätigung dieses Konzepts konnten ihnen Kollegen aus der Universität Würzburg liefern. Für die dafür notwendige Technik, die Photoelektronenspektroskopie (PES), sind Professor Friedrich Reinert, Inhaber des Lehrstuhls für Experimentelle Physik VII, und sein Mitarbeiter Privatdozent Dr. Achim Schöll schon seit Längerem Spezialisten.
Das Würzburger Experiment
„Wir bestrahlen die jeweilige Probe mit UV-Licht einer bestimmten Wellenlänge“, beschreibt Schöll die experimentelle Vorgehensweise. Das UV-Licht ist in der Lage, Elektronen aus den äußeren Hüllen zu lösen; die Analyse des Winkels, in dem die Elektronen ihre Bahn verlassen, ermöglicht den Physikern Rückschlüsse auf die Form der jeweiligen Orbitale. „Wir können auf diese Weise die Struktur eines Molekülorbitals darstellen und damit die richtige Theorie herausfinden“, sagt Schöll.
Was so einfach klingt, ist in Wirklichkeit experimentell äußert aufwendig. „Wir verdampfen dafür die Moleküle in einem extremen guten Vakuum“, beschreibt Schöll seine Arbeit. Anschließend schlägt der Dampf auf einer Oberfläche nieder wie Luftfeuchtigkeit auf dem Badezimmerspiegel. Mit dem Unterschied, dass es sich bei der Oberfläche im Labor um eine sehr spezielle handelt: „Sie besteht aus einem Einkristall und ist so glatt wie es überhaupt nur möglich ist“, so der Physiker. Dort lagern sich die Moleküle in nur einer einzigen Lage an – absolut flach liegend und alle mit gleicher Blickrichtung. Nur so ist garantiert, dass die Messung der Winkelabhängigkeit ein unverfälschtes Ergebnis liefert.
Die Bayreuther Theorie
Für die Entwicklung der passenden Theorie war die Bayreuther Arbeitsgruppe um Professor Stephan Kümmel verantwortlich. Die Physiker dort haben – im Rahmen der Dichtefunktionaltheorie – ein Konzept entwickelt, das es erlaubt, Molekülorbitale mit hoher Genauigkeit zu berechnen. Kümmels Mitarbeiter Matthias Dauth hat dieses Verfahren auf spezielle Moleküle organischer Halbleiter angewendet. Anschließend hat er die Ergebnisse mit Berechnungen verglichen, zu denen die physikalische Forschung von anderen theoretischen Ansätzen aus gelangt. „Die Unterschiede waren signifikant“, berichtet Dauth. „Um nachzuweisen, dass das in Bayreuth entwickelte Berechnungsverfahren die präziseren Vorhersagen erlaubt, war daher der Vergleich mit experimentellen Ergebnissen, also möglichst leistungsstarken spektroskopischen Untersuchungen, ausgesprochen wichtig.“
„Diese Übereinstimmung von Theorie und Experiment ermutigt uns, das theoretische Konzept weiterzuentwickeln, um die Elektroneneigenschaften noch genauer bestimmen zu können“, erklärt Kümmel. „Wir gewinnen auf diese Weise direkten Einblick in die elektronischen Eigenschaften von Materialien, die sich für neue Halbleitertechnologien mit großem Gewinn nutzen lassen, nicht zuletzt bei der Entwicklung effizienter Verfahren der Stromerzeugung.“
M. Dauth, T. Körzdörfer, and S. Kümmel; J. Ziroff, M. Wiessner, A. Schöll, and F. Reinert; M. Arita and K. Shimada: Orbital Density Reconstruction for Molecules, Physical Review Letters doi: 10.1103/PhysRevLett.107.193002
Kontakt
PD Dr. Achim Schöll, T: (0931) 31-85127, E-Mail:
![]() achim.schoell@physik.uni-wuerzburg.de
achim.schoell@physik.uni-wuerzburg.de